Note that even though row ids of the sample table should contain all column names of the expression matrix, they do not need to be in the same order.
Now we perform the projection using “Projection” >> “Project bulk data” function on the atlas page, where it asks to provide these two files, together with a name which will be used as a prefix to the projected samples for easier identification (we use “hp” as the name in this example). We also leave the sample column field blank here, which indicates we will use the first column of the sample table to group the projected samples together.
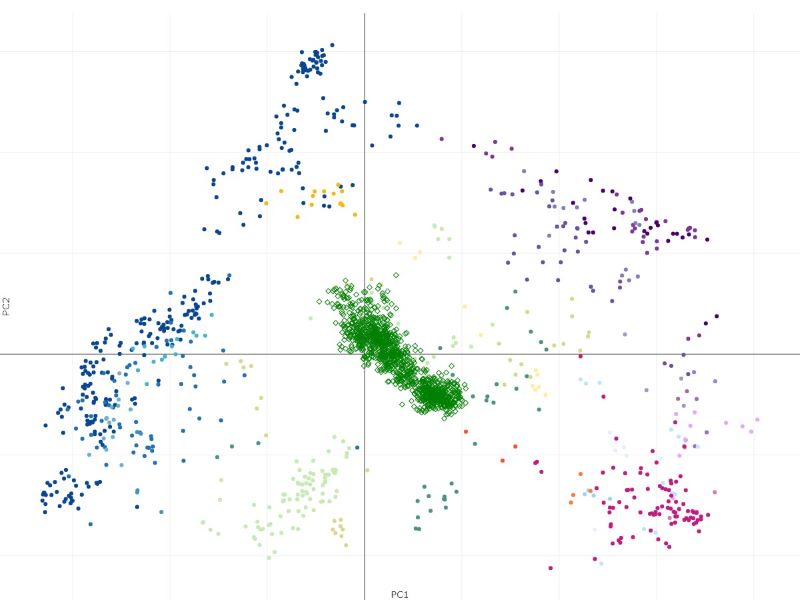
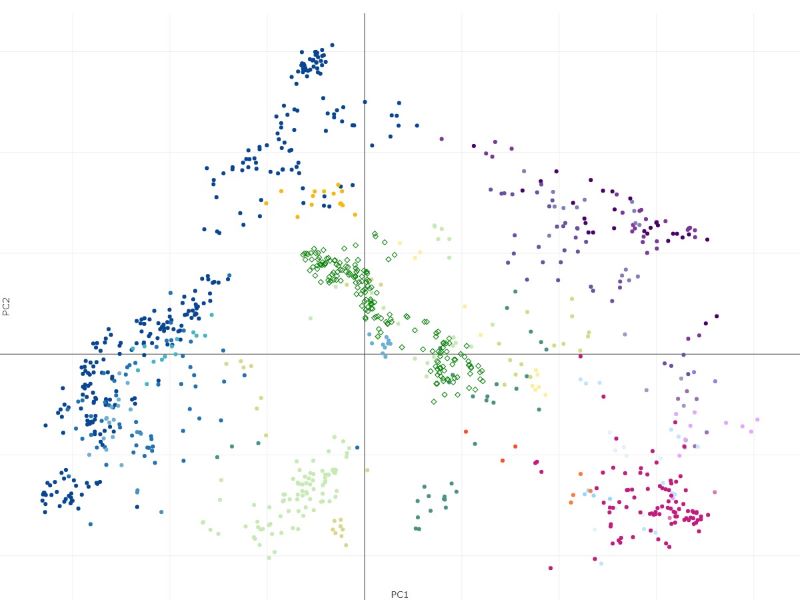
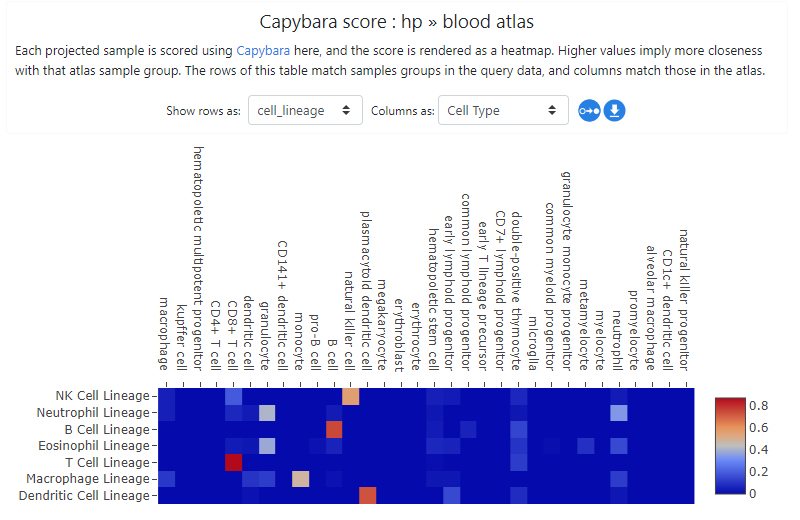
The projection process may take some time - it will scale with the expression file size being uploaded. The result is obtained as Capybara scores (Kong et.al.). In this example, the resulting projection shows a good biological concordance between the projected samples and cell types in the atlas, giving us some confidence that projection has worked well. Clicking on the "Show projections on PCA plot" button or simply going to Sample groups tab will also show the projected samples in the PCA space.
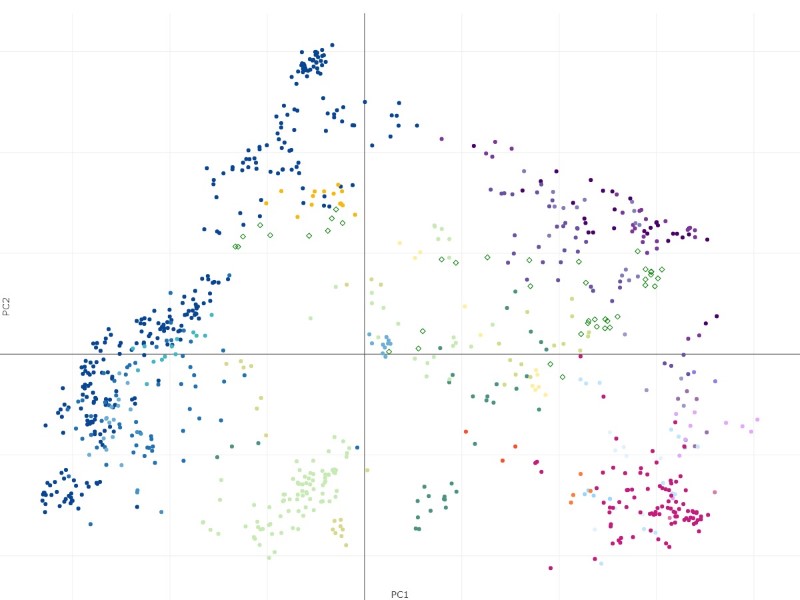
The samples from Haemopedia-Human-RNASeq data (diamond shapes) project onto the Stemformatics Blood Atlas near expected cell types. It is easier to see this by using the atlas page directly for projection using the files downloaded.
Projecting single cell data¶
Single cell RNA-seq data can be projected onto the atlas, but we have found that aggregating the data beforehand yields much better results than naively projecting each sample onto the atlas. This is due to the difference in the data structure between bulk and single cell RNA-seq data. There will also be performance issues for very large single cell datasets, both in terms of file upload and for processing if aggregation is not done.
As an example, we take this study by Villani, et. al. where they profiled human dendritic cells and monocytes to find sub clusters. We can download the files easily through the Broad Institute’s Single Cell Data Portal (note - you need to register and sign-in before downloading). We download “expression_matrix_tpm.txt” and “metadata.txt” files.
Note that the expression matrix here has gene symbols, rather than Ensembl gene ids, so we need to convert these first before projecting onto the atlas. There are various ways of obtaining the mapping between Ensembl gene ids to gene symbols, including using biomart or mygene.info. One simple option is to simply use the file provided on the atlas page :"Tools" >> "download data" >> "genes matrix", which contains this mapping.